 資源預覽
資源預覽



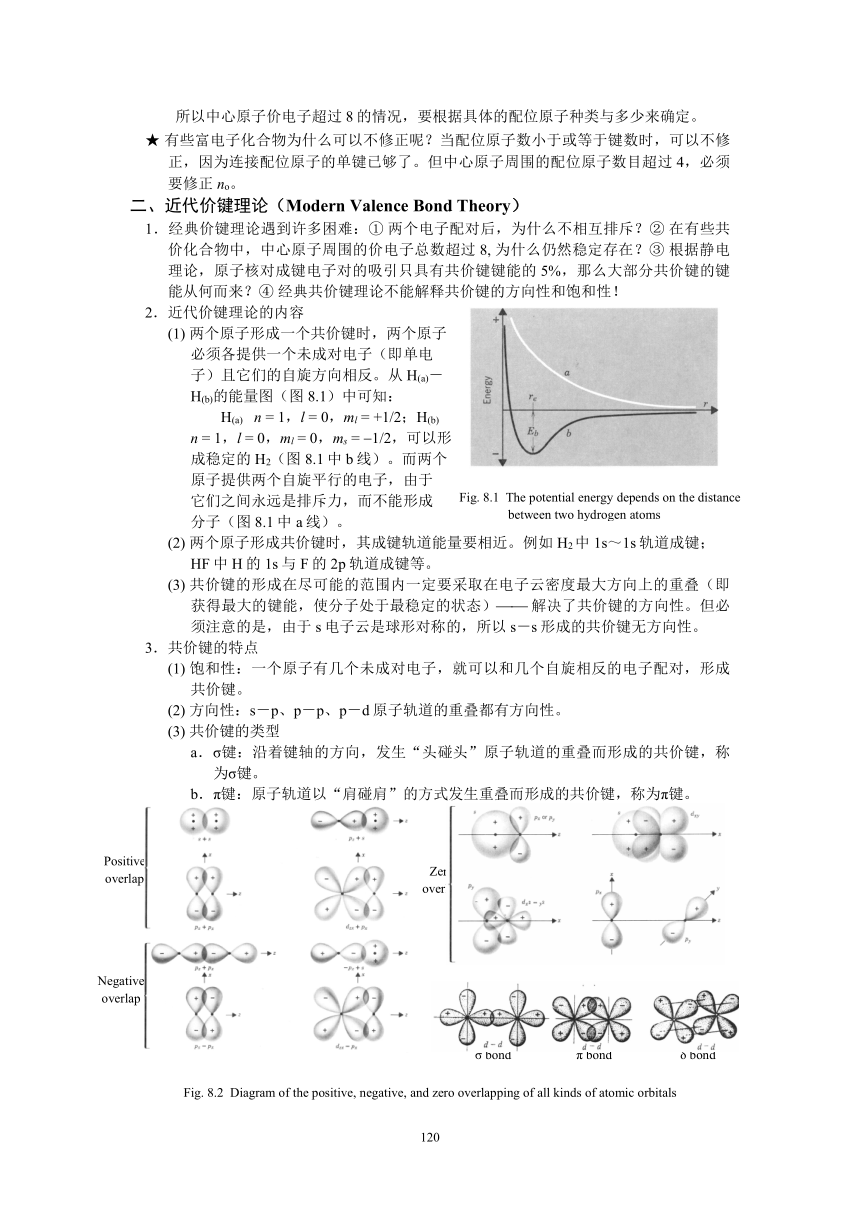

資源預覽